





서론
대식세포활성증후군(macrophage activation syndrome, MAS)은 이차성 혈구탐식림프조직구증(hemophagocytic lymphohistiocytosis, HLH)의 한 형태로, 조절을 벗어난 T 세포와 대식세포에 의해 발생하는 과염증(hyperinflammatory) 현상이다[1]. MAS는 지속되는 발열, 간비종대(hepatosplenomegaly), 혈구감소증 및 다장기부전(multi-organ dysfunction)을 특징으로 하며, 실제 임상에서 감염, 악성종양 및 류마티스 질환의 합병증 양상으로 마주칠 수 있다[2,3]. 전신형 소아기특발성관절염(systemic juvenile idiopathic arthritis, SJIA) 환자의 ~10%에서 MAS가 발생되며, ~5%의 전신홍반루프스(systemic lupus erythematosus, SLE) 환자와 ~2%의 가와사키병(Kawasaki disease, KD) 환자에서도 MAS의 발생이 보고되었다[4,5].
MAS는 사망률 8%–22%의 치명적 질병으로, 조기에 인식하여 적극적 치료를 하는 것이 매우 중요하다[1–3]. 하지만 발열, 혈소판감소증, 간 기능장애와 같은 MAS의 주요한 특징은 JIA, SLE 및 KD와 같은 기저질환(underlying disease)이 악화될 때에도 관찰되기 때문에, 진단적 혼란을 초래하기도 한다[5,6]. 특히 알려진 기저질환 없이 초기 임상양상이 MAS인 경우, 기저질환의 존재가 가려질 수 있다[7]. 본 연구에서 저자들은 초기 임상양상이 ‘MAS가 합병된 KD (MAS complicating KD, MAS-KD)’이었던 SJIA 증례를 경험하여 문헌고찰과 함께 보고한다.
증례
16개월 여아가 5일 동안의 발열로 입원하였다. 출생력과 가족력에는 특이 병력이 없었으나 생후 6개월 다른 대학병원에 불명열(fever of unknown origin, FUO)로 치료받았던 과거력이 있다. 당시 4주 동안 반복되는 발열과 발진으로 입원하였고 빈혈, 혈소판감소증, 간효소치 상승이 확인되었으나 특별한 치료 없이 발열과 검사실 이상이 회복되어 퇴원하였다. 환자의 신장은 79 cm(50–75th percentile), 몸무게는 9.2 kg(15–25th percentile)였다.
입원 시 활력 증후는 혈압 90/60 mmHg, 심박수 134회/분, 호흡 24회/분, 체온 37.2℃이었다. 신체 진찰에서 간(3 cm)과 비장(5 cm)의 촉진 외에 비정상 소견은 없었다. 혈액검사에서 혈색소 11.6 g/dL, 백혈구 15,700 /μL (호중구 72%), 혈소판 391,000 /μL이었고 적혈구 침강속도(erythrocyte sedimentation rate, ESR) 48 mm/hr, C-반응단백(C-reactive protein, CRP) 52 mg/L (정상 < 5.0 mg/L), 알부민 3.3 g/dL, aspartate aminotransferase(AST) 29 U/L, alanine aminotransferase(ALT) 14 U/L이었으며, 류마티스 인자(rheumatoid factor, RF)와 자가항체(antinuclear antibody, ANA)는 음성이었다. 소변검사와 흉부 영상의학 검사도 정상이었다. 세균감염 가능성(백혈구증가증, ESR 및 CRP 상승)을 배제할 수 없어 경험적 항생제(ampicillin-sulbactam) 치료를 시작하였다. 하지만 입원 5일째에도 발열은 조절되지 않았다. 뇌척수액 검사와 미생물학적 검사(배양검사, polymerase chain reaction [PCR], 특이 항체검사)에서 주목할 만한 소견은 없었다. 심장초음파 검사에서 관상동맥 이상(coronary artery abnormalities, CAAs)과 같은 특이 병변은 관찰되지 않았고, 복부 전산화 단층촬영(computed tomography, CT)에서 간비종대를 제외하고 뼈 스캔(bone scan)을 포함한 다른 영상의학 검사도 정상이었다.
입원 7일째, 하루 2–3회 38℃ 이상 발열이 측정되었고 추적 혈액검사 소견도 악화되었다. 혈색소 9.2 g/dL, ESR 56 mm/hr, CRP 184 mg/L, 알부민 2.7 g/dL, AST 199 U/L, ALT 109 U/L. 또한 전날까지 관찰되지 않았던 두드러기 양상의 발진, 양안의 충혈, 입술의 홍조와 균열이 나타났다. 지속되는 발열, 점막피부 증상 및 검사실 소견(빈혈, 저알부민혈증, 간효소치 상승 및 백혈구증가증)을 토대로, 불완전(incomplete) KD를 진단하고 정맥주사 면역글로불린(intravenous immunoglobulin [IVIG], 2 g/kg/dose)과 아스피린 치료를 추가하였다. IVIG 치료 후 임상양상이 일시적으로 호전되는 것처럼 보였지만 다시 발열이 발생되어 정맥주사 스테로이드(intravenous methylprednisolone [IVMP], 30 mg/kg/day for 3 days)를 시행하였다(Fig. 1).
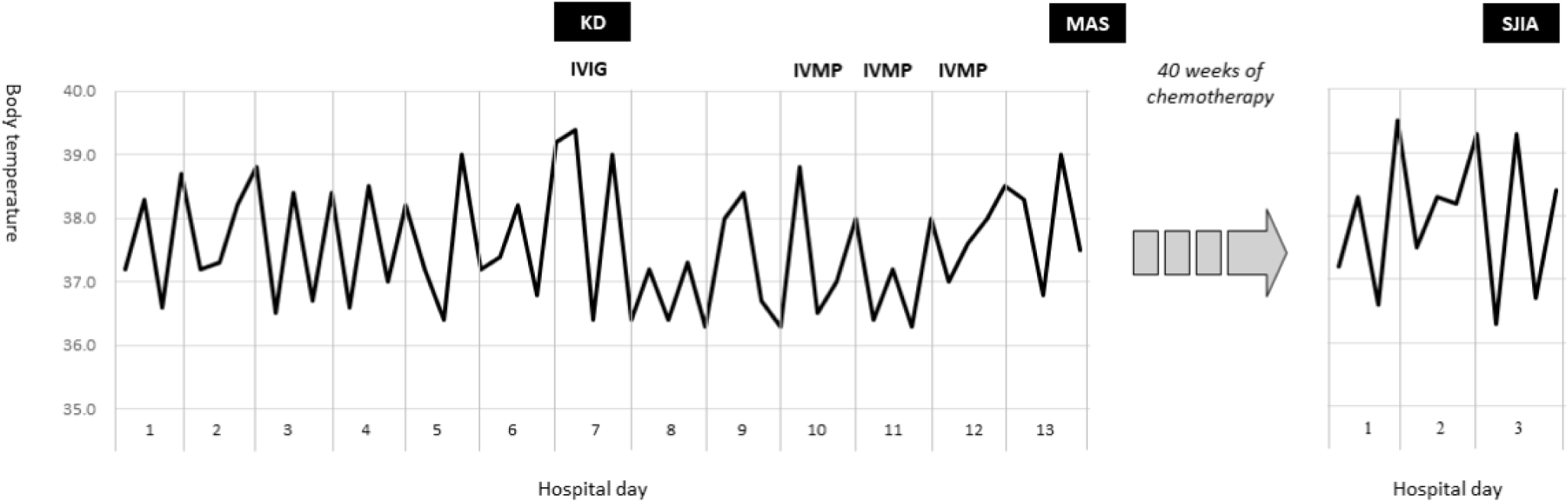
입원 14병일째 검사실 소견은 혈색소 6.4 g/dL, 백혈구 3,800 /μL (호중구 63%), 혈소판 51,000 /μL, AST 1,006 U/L, ALT 397 U/L, 중성지방 197 mg/dL, 섬유소원(fibrinogen) 139 mg/dL, 페리틴(ferritin) 21,900 ng/mL으로 좀 더 악화되었다. 골수 생검에서는 전형적인 혈구탐식구증(hemophagocytosis)이 관찰되지는 않았으나 다른 검사실 소견은 HLH 혹은 MAS 진단기준(Table 1)[8–10]을 만족하여, 덱사메타손(dexamethasone, 10 mg/m2)과 에토포시드(etoposide, 150 mg/m2), 사이클로스포린(cyclosporine, 6 mg/kg/day)을 포함하는 HLH-2004 치료지침(therapeutic protocol)[8]에 따라 복합 화학요법 치료를 시행하였다. 치료 기간 40주 동안 급성 세기관지염으로 한차례 입원한 것을 제외하고는 질병 활성도가 잘 조절되어 환아는 생후 28개월에 완전 관해(complete remission) 판정을 받았다.
1) Patient in this study met five criteria of the six HLH-2004 criteria tested (i.e., sCD25 levels and NK cell activity were not tested) and all 2016 MAS-SJIA criteria. The patient’s HScore was 264 points.
HLH: hemophagocytic lymphohistiocytosis; MAS: macrophage activation syndrome; SJIA: systemic juvenile idiopathic arthritis; 2016 MAS-SJIA: the 2016 consensus criteria for MAS complicating SJIA; PRINTO: Paediatric Rheumatology International Trials Organisation; HScore, hemophagocytic syndrome diagnostic score; SM: splenomegaly; HM: hepatomegaly; HSM: hepatosplenomegaly; CBC: complete blood cell count; AST: aspartate aminotransferase; TG: triglycerides; sIL-2R: soluble interleukin-2 receptor; NK: natural killer; BM: bone marrow.
치료 종결 2개월 후, 환아는 발열과 발진, 관절통으로 다시 입원하였다. 간비종대와 백혈구증가증(18,500 /μL)을 보였으나 관절 부종이나 열감은 없었고 뼈 스캔도 정상이었다. 그러나 환아는 양측 무릎과 발목의 통증으로 걸을 수 없었고 밤에도 심하게 보채는 증상을 보였다. 즉, 환아의 임상양상은 SJIA의 진단기준에 부합하였다(Table 2)[11,12]. 단기간의 경구 스테로이드(prednisone 1 mg/kg for 7 days) 치료와 함께 임상양상은 빠르게 호전되었다. 환아는 퇴원 후 외래에서 SJIA에 대한 유지 치료(methotrexate와 naproxen)를 받았고 추적기간 동안 MAS-KD의 재발이나 SJIA의 악화는 없었다.
고찰
본 증례 환아의 초기 임상양상은 조절되지 않은 발열과 심한 전신염증이었다. 입원 7일째 점막피부 증상을 비롯한 KD의 주요한 증상(principal features)이 나타났고 적절한 치료에도 임상양상은 악화되어 입원 14일째 MAS의 진단기준을 충족하였다. 40주 동안 화학요법 중에는 질병의 활성도가 잘 조절되었지만, 관해 판정 후 면역억제 치료(immunosuppressive therapy)가 중단됨으로써 전형적인 SJIA의 증상이 나타난 것으로 여겨진다. 본 환아와 같은 복잡한 임상 시나리오에서 MAS와 기저질환을 조기에 인식하기 위해서는 (1) MAS의 임상양상, (2) MAS-KD의 특징 그리고 (3) KD와 SJIA의 관계를 이해하는 것이 필요하다.
MAS는 심한 전신염증을 유발할 수 있는 여러 종류의 소아 질환에서 발생되는 드물지만 치명적인 합병증이다. 과거에는 MAS는 ‘류마티스 연관(rheumatic disease-associated) 이차성 HLH’에 국한된 개념이었으나, 최근에는 류마티스 질환 외에 다양한 의학적 상황에서 MAS가 보고되어 많은 전문가들은 MAS를 이차성 HLH와 동일한 의미로 사용하기도 한다[3]. MAS의 핵심적인 병리기전은 조절을 벗어난 면역 세포들에 의한 사이토카인 폭풍(cytokine storm) 현상이다[1,2]. 과생산된 전염증성(proinflammatory) 사이토카인은 발열, 간비종대, 혈구감소증, 고페리틴혈증, 혈구탐식구증과 같은 심한 전신염증 소견과 신경학적, 혈액학적 이상 등, 주요 장기의 기능부전을 유발시킨다[4]. 하지만 이러한 임상양상은 MAS에만 나타나는 특징(pathognomonic)이 아니라 패혈증과 같은 심한 감염, 악성종양 및 류마티스 질환 악화 시에도 관찰되기 때문에, 실제 임상에서 MAS의 발생을 제대로 인식하지 못할 수 있다[5,6]. 따라서 적절한 치료에도 불구하고, 설명되지 않는 발열, 간비종대, 혈소판감소증, 간효소치 상승, 혹은 고페리틴혈증을 나타내는 환자에서 MAS의 동반 가능성을 임상적으로 의심하는 것이 중요하다[9,13].
본 증례 환자의 초기 임상양상은 ‘MAS가 합병된 KD (MAS-KD)’였다. García-Pavón et al.[14]은 체계적 검토(systematic review)를 통해 69명 MAS-KD 환자의 자료를 분석하였다. 환자의 평균 연령은 5.6세였고 남자(68%)와 아시아 인종(75%)이 흔했다. 모든 환자는 발열(100%)을 보였고 비종대(splenomegaly)는 69%에서 관찰되었다. 대부분 환자에서 혈소판감소증(87%)과 간효소치 상승(94%) 및 고페리틴혈증(95%)이 확인되었다. 또한, IVIG 저항성(90%)과 CAAs(46%)의 비율도 높았다. 환자의 61%는 단기간의 면역조절제 치료를 받았고, 나머지 39% 환자는 HLH-2004 치료지침에 따라 에토포시드와 사이클로스포린을 포함하는 복합 화학요법을 시행받았다. MAS-KD의 사망률은 13%로, MAS가 합병된 SJIA(MAS complicating SJIA, MAS-SJIA)와 유사했다[2,4].
KD 환자에서 MAS가 발생될 때, 대부분(73%)은 본 증례의 환자와 같이 KD 진단 후 증상이 악화되어 MAS로 진행한다[5,6]. 하지만 약 20% 환자들은 KD와 MAS 증상을 동시에 나타내며, 일부 환자(6%)에서는 MAS 진단 후 뒤늦게 KD 증상을 보이기도 한다[14,15]. 따라서 KD나 SJIA와 같은 기저질환을 조기에 인식하기 위해서는 점막피부 병변이나 관절통 등 관련 증상 여부와 무관하게, MAS 환자에서 숨겨진 기저질환의 존재 가능성을 고려하여 임상경과를 세밀하게 추적 관찰해야 한다.
본 증례 환자의 최종 진단은 SJIA이다. SJIA는 소아 MAS의 가장 흔한 원인이며, 다수관절형(polyarticular) JIA나 소수관절형(pauciarticular) JIA와는 구분되는 특징을 나타내어 다른 아형의 JIA와는 별도의 질환으로 간주된다[15]. 실제로 자가면역(autoimmune) 질환인 다른 아형의 JIA와 달리, SJIA는 KD와 동일하게 자가염증(autoinflammatory) 질환으로 분류된다[12]. Han et al.[16]은 KD와 SJIA는 중증도의 차이를 보이는 하나의 임상 증후군으로, KD에 대한 유전적 감수성(genetic susceptibility)을 지닌 환자에서는 일회성, 단기간(short-term) 임상경과가 관찰되며 SJIA에 대한 유전적 감수성을 지닌 환자에서는 재발성, 장기간(long-term) 임상경과가 관찰될 것이라 제안하였다. 즉, 두 질환은 초기 임상양상이 완전히 동일할 수 있고 단기간의 임상경과는 KD로, 장기간의 임상경과는 SJIA로 구분할 수 있다는 것이다.
KD와 SJIA 연관성에 관한 Dong et al.[17]의 연구(n = 15)와 Go et al.[18]의 연구(n = 8)에서, ‘SJIA가 함께 진단된 KD (KD with a co-diagnosis of SJIA, KD/SJIA)’의 특징을 보고하였다. 전형적인 KD와 비해, KD/SJIA에는 결막염 증상이 없는 환자, 전신 림프절 비대나 간비종대가 동반된 환자, Z-score > 5.0의 CAAs가 없는 환자, 2회 이상 IVIG 치료를 받은 환자 및 MAS가 합병된 환자가 상대적으로 많았다[17,18]. 검사실 특징으로는, 저알부민혈증, 간효소치 상승 혹은 백혈구증가증을 나타내는 환자가 많았다[18]. 하지만 이러한 임상 및 검사실 소견은 KD/SJIA뿐만 아니라, 불완전 KD나 치료불응 KD 등 심한 형태의 KD에서도 관찰 가능하다는 것을 유념해야 한다.
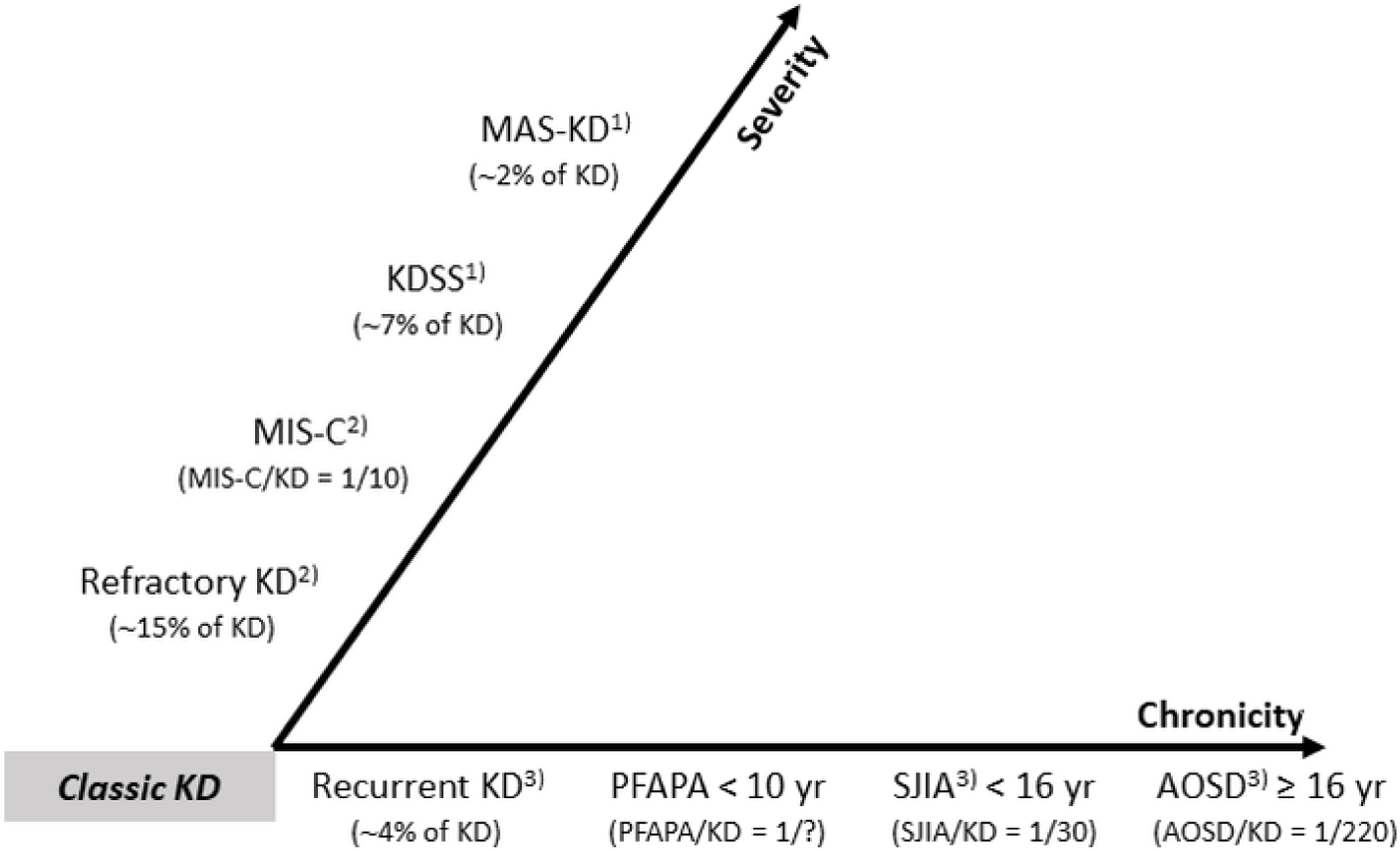
Fig. 2에는 KD와 KD-유사 증상(KD-like features)을 보일 수 있는 질환들을 중증도(severity)와 만성화(chronicity)에 따라 분류하였다[6,11,13,19–22]. 각각의 질환은, 병리기전이 다른, 독립적인 질환이지만 임상 표현형(clinical phenotypes)은 유사하거나 동일할 수 있다[21,22]. 코로나바이러스감염증-19(coronavirus disease 2019, COVID-19) 팬데믹 동안 KD-유사 증상을 나타내는 소아다기관염증증후군(multisystem inflammatory syndrome in children, MIS-C)이 KD 연구 분야에서 흥미로운 주제가 되었던 것처럼[13], KD와 SJIA의 관계를 이해하는 것은 KD-유사 증상을 나타낼 수 있는 다양한 소아 질환의 연구에 유용한 단서를 제공할 것이다.
결론
KD는 소아 MAS의 주요한 원인 중 하나로, 적절한 치료에도 임상적 호전이 없는 KD 환자에서 MAS 동반 가능성을 의심해야 한다. 동시에 MAS는 심한 전신염증을 유발하는 다양한 의학적 상황에 합병되는 과염증 현상이므로, 숨겨진 기저질환의 존재 가능성도 함께 고려해야 한다. 본 연구에서 저자들은 초기 임상양상이 불완전 KD와 MAS이었던 SJIA 환아의 증례를 기술하였다. KD, MAS 및 SJIA는 독립적 질병이지만, 유사한 임상 표현형을 공유한다. 이 세 가지 질병의 관계는 KD 연구 분야의 흥미로운 주제가 될 것이다.